|
|
|
|

|
|
|
|
LORBRENA is indicated for the treatment of adult patients with metastatic non-small cell lung cancer
(NSCLC) whose tumors are anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved
test.
|
|
|
|
|
Important Safety Information
|
|
Contraindications: LORBRENA
is contraindicated in patients taking strong CYP3A inducers, due to the potential for serious
hepatotoxicity.
Risk
of Serious Hepatotoxicity with Concomitant Use of Strong CYP3A Inducers:
Severe
hepatotoxicity occurred in 10 of 12 healthy subjects receiving a single dose of LORBRENA with
multiple daily doses of rifampin, a strong CYP3A inducer. Grade 4 ALT or AST elevations occurred in
50% of subjects, Grade 3 in 33% of subjects, and Grade 2 in 8% of subjects. ALT or AST elevations
occurred within 3 days and returned to within normal limits after a median of 15 days (7 to 34
days); median time to recovery in subjects with Grade 3 or 4 or Grade 2 ALT or AST elevations was 18
days and 7 days, respectively. LORBRENA is contraindicated in patients taking strong CYP3A inducers.
Discontinue strong CYP3A inducers for 3 plasma half-lives of the strong CYP3A inducer prior to
initiating LORBRENA.
Please
scroll down for additional Important Safety Information.
|
|
|
|
|
Dear Healthcare Provider,
Review the PFS results from the primary and latest follow-up analyses for
LORBRENA. See how the CROWN data may help inform your first-line treatment decision for your next patient
with ALK+ mNSCLC.1,2
Study
design: CROWN is a global, open-label, randomized, multicenter,
Phase 3 trial in which 296 adults with previously untreated ALK-positive locally advanced or metastatic NSCLC were randomized 1:1 to receive
LORBRENA 100 mg QD (n=149) or crizotinib 250 mg BID (n=147). The major efficacy outcome measure of the
trial is PFS based on BICR. Additional efficacy outcome measures included ORR, DoR, IC-ORR, IC-DoR, and
OS.1-3
|
|
|
|
In the primary
analysis of the first-line ALK+ mNSCLC CROWN
trial
|
|
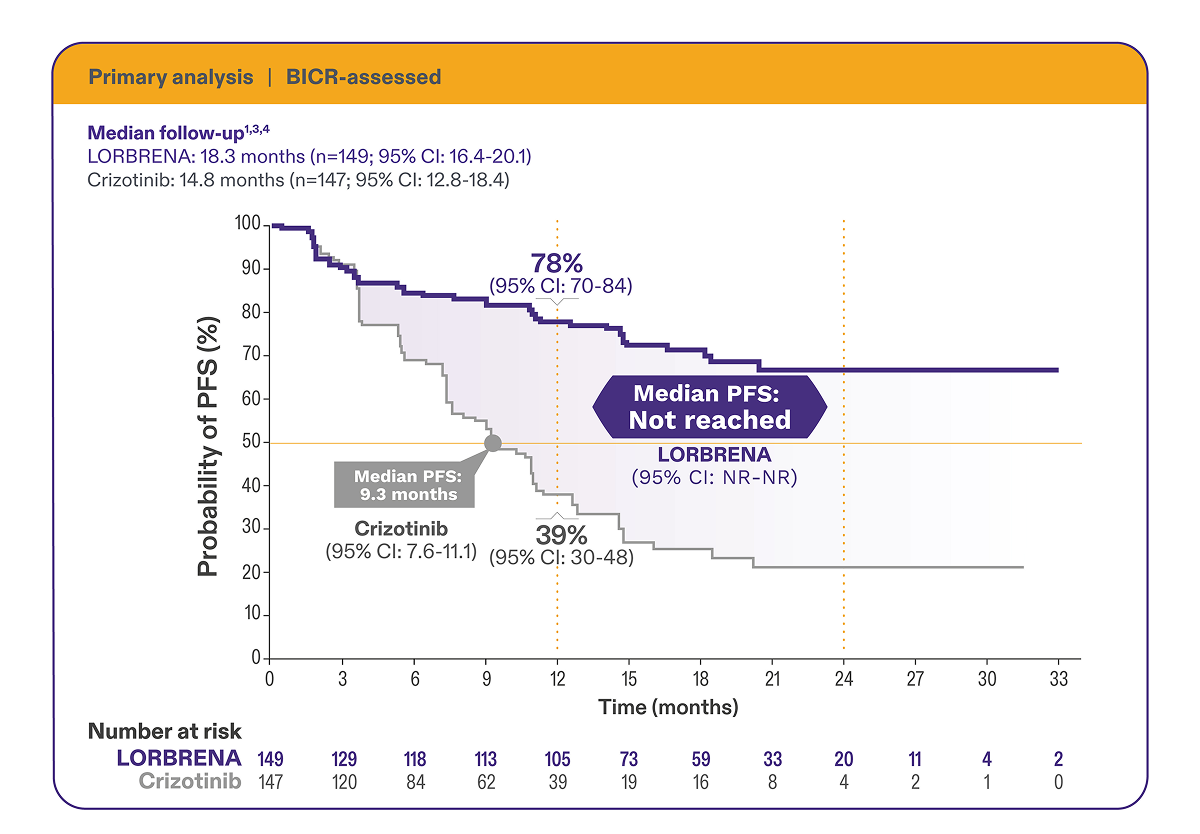
Superior progression-free survival vs crizotinib1,3
|

|
|
|
•
|
mPFS
was not reached for LORBRENA vs 9.3 months (95% CI: 7.6-11.1)
for crizotinib at the 18.3-month follow-up (HR=0.28 [95% CI: 0.19-0.41]; P<0.0001)1,3
|
|
•
|
At
the data cutoff, OS data were not mature1
|
|
•
|
ORR† for LORBRENA was 76% (95% CI: 68-83) vs 58% (95% CI: 49-66) for
crizotinib3
|
•
|
Response duration ≥12 months for LORBRENA was 70% vs 27% for
crizotinib1
|
|
•
|
For LORBRENA, 73% partial response and 3% complete response; for
crizotinib, 58% partial response and 0% complete response3
|
|
|
•
|
IC-ORR†‡ for LORBRENA was 82% (95% CI: 57-96) vs 23% (95% CI: 5-54) for
crizotinib3,4§
|
•
|
Response duration ≥12 months for LORBRENA was 79% vs 0% for
crizotinib1
|
|
•
|
For LORBRENA, 12% partial response and 71% complete response;
for
crizotinib, 15% partial response and 8% complete response3,4§
|
|
|
|
*
|
Based on 1-sided stratified log-rank test.1
|
|
|
†
|
Assessed by BICR per RECIST v1.1.1,3
|
|
|
‡
|
Intracranial response and DoR were evaluated in a prespecified exploratory analysis of 30
patients with measurable CNS lesions at baseline.1
|
|
|
|
|
|
|
In an
exploratory analysis of the first-line ALK+ mNSCLC CROWN
trial
|
|
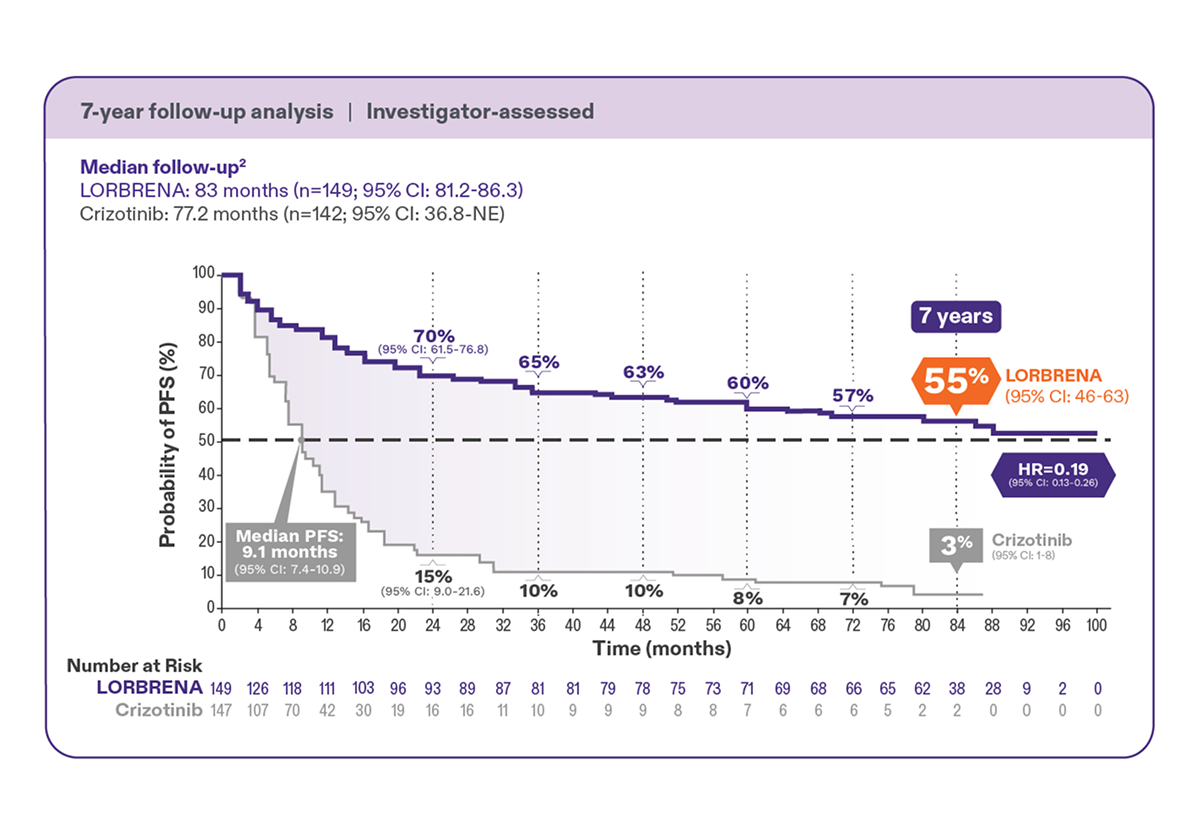
Median progression-free survival was not reached at 83 months (95% CI: 68.5-NR)2
|

|
|
|
•
|
Patients on LORBRENA had a 55% probability of being alive and
progression free at 7 years2
|
|
•
|
81% reduction in risk of progression or death vs crizotinib
(investigator-assessed) (HR=0.19 [95% CI: 0.13-0.26])2
|
|
•
|
At the data cutoff, OS data were not mature2
|
|
|
|
|
LORBRENA has an established safety profile2
|
Adverse
reactions in the CROWN trial1
|
•
|
Serious
adverse reactions occurred in 34% of the 149 patients treated
with LORBRENA; the most frequently reported serious adverse reactions were pneumonia (4.7%), dyspnea
(2.7%), respiratory failure (2.7%), cognitive effects (2.0%), and pyrexia (2.0%). Fatal adverse
reactions occurred in 3.4% of patients and included pneumonia (0.7%), respiratory failure (0.7%),
cardiac failure acute (0.7%), pulmonary embolism (0.7%), and sudden death (0.7%)
|
|
•
|
The
most frequently reported (≥20%) adverse reactions for
LORBRENA
and crizotinib, respectively, were edema (56% vs 40%; 4% vs 1.4% [Grade 3 or 4]), weight gain (38%
vs
13%; 17% vs 2.1%); peripheral neuropathy (34% vs 15%; 2% vs 0.7%), cognitive effects (21% vs 6%; 2%
vs
0%), diarrhea (21% vs 52%; 1.3% vs 0.7%), and dyspnea (20% vs 16%; 2.7% vs 2.1%)
|
|
•
|
The
most frequent laboratory abnormalities for LORBRENA and
crizotinib, respectively, were hypertriglyceridemia (95% vs 27%; 22% vs 0% [Grade 3 or 4]),
hypercholesterolemia (91% vs 12%; 19% vs 0%), increased creatinine (81% vs 99%; 0.7% vs 2.1%),
increased GGT (52% vs 41%; 6% vs 6%), hyperglycemia (48% vs 27%; 7% vs 2.1%), anemia (48% vs 38%; 2%
vs 2.8%), increased AST (48% vs 75%; 2% vs 3.5%), increased ALT (44% vs 75%; 2.7% vs 4.3%),
increased
CPK (39% vs 64%; 2% vs 5%), and hypoalbuminemia (36% vs 61%; 0.7% vs 6%)
|
|
•
|
Median
duration of exposure to LORBRENA was 16.7 months (range, 4
days
to 34.3 months); 76% of patients received LORBRENA for at least 12 months
|
|

|
|
|
|
Therapy management strategies
|
|
Discussing ARs with patients and their care partners prior to and during
treatment with LORBRENA is important for appropriate therapy management.5
When your patients experience ARs, there are ways to monitor,
assess, and help manage ARs to support patients on LORBRENA.5
Explore
more about therapy management strategies for specific adverse reactions, including dose
modifications.1
|

|
|
|
|
IMPORTANT SAFETY INFORMATION (CONT'D)
|
|
|
Central Nervous System (CNS) Effects: A broad spectrum of CNS effects can occur;
overall, CNS effects occurred in 52% of the 476 patients receiving LORBRENA. These included seizures
(1.9%, sometimes in conjunction with other neurologic findings), psychotic effects (7%; 0.6% severe
[Grade 3 or 4]), and changes in cognitive function (28%; 2.9% severe), mood (including suicidal
ideation) (21%; 1.7% severe), speech (11%; 0.6% severe), mental status (1.3%; 1.1% severe), and sleep
(12%). Median time to first onset of any CNS effect was 1.4 months (1 day to 3.4 years). Overall, 2.1%
and 10% of patients required permanent or temporary discontinuation of LORBRENA, respectively, for a CNS
effect; 8% required dose reduction. Withhold and resume at same or reduced dose or permanently
discontinue based on severity.
|
|
Hyperlipidemia: Increases in serum cholesterol and triglycerides can occur. Grade 3 or
4 elevations in total cholesterol occurred in 18% and Grade 3 or 4 elevations in triglycerides occurred
in 19% of the 476 patients who received LORBRENA. Median time to onset was 15 days for both
hypercholesterolemia and hypertriglyceridemia. Approximately 4% and 7% of patients required temporary
discontinuation and 1% and 3% of patients required dose reduction of LORBRENA for elevations in
cholesterol and in triglycerides in Study B7461001 and Study B7461006, respectively. Eighty-three
percent of patients required initiation of lipid-lowering medications, with a median time to onset of
start of such medications of 17 days. Initiate or increase the dose of lipid-lowering agents in patients
with hyperlipidemia. Monitor serum cholesterol and triglycerides before initiating LORBRENA, 1 and 2
months after initiating LORBRENA, and periodically thereafter. Withhold and resume at same dose for the
first occurrence; resume at same or reduced dose of LORBRENA for recurrence based on severity.
|
|
Atrioventricular (AV) Block: PR interval prolongation and AV block can occur. In 476
patients who received LORBRENA at a dose of 100 mg orally once daily and who had a baseline
electrocardiography (ECG), 1.9% experienced AV block and 0.2% experienced Grade 3 AV block and underwent
pacemaker placement. Monitor ECG prior to initiating LORBRENA and periodically thereafter. Withhold and
resume at reduced or same dose in patients who undergo pacemaker placement. Permanently discontinue for
recurrence in patients without a pacemaker.
|
|
Interstitial Lung Disease (ILD)/Pneumonitis: Severe or life-threatening pulmonary
adverse reactions consistent with ILD/pneumonitis can occur. ILD/pneumonitis occurred in 1.9% of
patients, including Grade 3 or 4 ILD/pneumonitis in 0.6% of patients. Four patients (0.8%) discontinued
LORBRENA for ILD/pneumonitis. Promptly investigate for ILD/pneumonitis in any patient who presents with
worsening of respiratory symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, and fever).
Immediately withhold LORBRENA in patients with suspected ILD/pneumonitis. Permanently discontinue
LORBRENA for treatment-related ILD/pneumonitis of any severity.
|
|
Hypertension: Hypertension can occur. Hypertension occurred in 13% of patients,
including Grade 3 or 4 in 6% of patients. Median time to onset of hypertension was 6.4 months (1 day to
2.8 years), and 2.3% of patients temporarily discontinued LORBRENA for hypertension. Control blood
pressure prior to initiating LORBRENA. Monitor blood pressure after 2 weeks and at least monthly
thereafter. Withhold and resume at reduced dose or permanently discontinue based on severity.
|
|
Hyperglycemia: Hyperglycemia can occur. Hyperglycemia occurred in 9% of patients,
including Grade 3 or 4 in 3.2% of patients. Median time to onset of hyperglycemia was 4.8 months (1 day
to 2.9 years), and 0.8% of patients temporarily discontinued LORBRENA for hyperglycemia. Assess fasting
serum glucose prior to initiating LORBRENA and monitor periodically thereafter. Withhold and resume at
reduced dose or permanently discontinue based on severity.
|
|
Embryo-fetal Toxicity: LORBRENA can cause fetal harm. Advise pregnant women of the
potential risk to a fetus. Advise females of reproductive potential to use an effective non-hormonal
method of contraception, since LORBRENA can render hormonal contraceptives ineffective, during treatment
with LORBRENA and for at least 6 months after the final dose. Advise males with female partners of
reproductive potential to use effective contraception during treatment with LORBRENA and for 3 months
after the final dose.
|
|
Adverse Reactions: In the pooled safety population of 476 patients who received 100 mg
LORBRENA once daily, the most frequent (≥ 20%) adverse reactions were edema (56%), peripheral neuropathy
(44%), weight gain (31%), cognitive effects (28%), fatigue (27%), dyspnea (27%), arthralgia (24%),
diarrhea (23%), mood effects (21%), and cough (21%). The most frequent (≥ 20%) Grade 3-4 laboratory
abnormalities in patients receiving LORBRENA were hypercholesterolemia (21%) and hypertriglyceridemia
(21%).
|
|
In previously untreated patients, serious adverse reactions occurred in 34% of the 149 patients treated
with LORBRENA; the most frequently reported serious adverse reactions were pneumonia (4.7%), dyspnea
(2.7%), respiratory failure (2.7%), cognitive effects (2.0%), and pyrexia (2.0%). Fatal adverse
reactions occurred in 3.4% of patients and included pneumonia (0.7%), respiratory failure (0.7%),
cardiac failure acute (0.7%), pulmonary embolism (0.7%), and sudden death (0.7%). In the Phase 1/2
study, serious adverse reactions occurred in 32% of the 295 patients; the most frequently reported
serious adverse reactions were pneumonia (3.4%), dyspnea (2.7%), pyrexia (2%), mental status changes
(1.4%), and respiratory failure (1.4%). Fatal adverse reactions occurred in 2.7% of patients and
included pneumonia (0.7%), myocardial infarction (0.7%), acute pulmonary edema (0.3%), embolism (0.3%),
peripheral artery occlusion (0.3%), and respiratory distress (0.3%).
|
|
Drug Interactions: LORBRENA is contraindicated in patients taking strong CYP3A
inducers. Avoid concomitant use with moderate CYP3A inducers, strong CYP3A inhibitors, and fluconazole.
If concomitant use of moderate CYP3A inducers cannot be avoided, increase the LORBRENA dose as
recommended. If concomitant use with a strong CYP3A inhibitor or fluconazole cannot be avoided, reduce
the LORBRENA dose as recommended. Avoid concomitant use of LORBRENA with CYP3A substrates and P-gp
substrates, which may reduce the efficacy of these substrates.
|
|
Lactation: Because of the potential for serious adverse reactions in breastfed infants,
instruct women not to breastfeed during treatment with LORBRENA and for 7 days after the final dose.
|
|
Hepatic Impairment: No dose adjustment is recommended for patients with mild (total
bilirubin ≤ ULN with AST > ULN or total bilirubin >1 to 1.5 × ULN with any AST) to moderate
(Child-Pugh B) hepatic impairment. In patients with severe hepatic impairment (Child-Pugh C), the
recommended dosage of LORBRENA is 50 mg orally once daily.
|
|
Renal Impairment: In patients with CLcr 15 to <30 mL/min (estimated by
Cockcroft-Gault), the recommended dosage of LORBRENA is 75 mg orally once daily. No dose adjustment is
recommended for patients with CLcr 30 to 89 mL/min (estimated by Cockcroft-Gault).
|
|
|
INDICATION
LORBRENA is indicated for the treatment of adult patients with metastatic
non-small cell lung cancer (NSCLC) whose tumors are anaplastic lymphoma kinase (ALK)-positive as detected
by an FDA-approved test.
Please
see full Prescribing
Information
for LORBRENA.
|
|
Information on FDA-approved tests for the detection of ALK rearrangements in NSCLC is available at http://www.fda.gov/CompanionDiagnostics.
|
ALK=anaplastic
lymphoma kinase; ALK+=anaplastic
lymphoma kinase–positive; ALT=alanine aminotransferase; AR=adverse reaction; AST=aspartate
aminotransferase; BICR=Blinded Independent Central Review; BID=twice daily; CI=confidence interval;
CNS=central nervous system; CPK=creatine phosphokinase; DoR=duration of response; FDA=US Food and Drug
Administration; GGT=gamma-glutamyl transferase; HR=hazard ratio; IC-DoR=intracranial duration of response;
IC-ORR=intracranial overall response rate; mNSCLC=metastatic non–small cell lung cancer; mPFS=median
progression-free survival; NE=not estimable; NR=not reached; NSCLC=non–small cell lung cancer; ORR=overall
response rate; OS=overall survival; PFS=progression-free survival; QD=once daily; RECIST=Response
Evaluation Criteria in Solid Tumors.
References:
| 1. |
LORBRENA® (lorlatinib)
Prescribing Information. Pfizer Inc; January 2026.
|
| 2. |
Shaw
AT, Solomon BJ, Felip E, et al. Lorlatinib versus crizotinib as first-line treatment for advanced
ALK-positive
non-small cell lung cancer: 7-year update from the phase 3 CROWN study. Ann
Oncol.
2026; doi: https://doi.org/10.1016/j.annonc.2026.05.692
|
| 3. |
Shaw
AT,
Bauer TM, de Marinis F, et al; CROWN Trial Investigators. First-line lorlatinib or crizotinib in
advanced ALK-positive
lung cancer. N
Engl J Med.
2020;383(21):2018-2029.
|
| 4. |
Data on file. Pfizer Inc, New York, NY.
|
| 5. |
Liu
G,
Mazieres J, Stratmann J, et al. A pragmatic guide for management of adverse events associated with
lorlatinib. Lung
Cancer.
2024;191:107535.
|
|
|
Connecticut
price disclosure information for prescribers available here.
Colorado
price disclosure information for prescribers available here.
Vermont
price disclosure information for prescribers available here.
Please
do not reply, as this email address cannot accept replies. If you wish to contact Pfizer,
click here.
View
the Pfizer Privacy Policy here.
Pfizer
P.O.
Box 29180
Mission, KS 66201
|

|
|
This
information is intended only for healthcare professionals in the United States.
Patients
should always ask their doctors for medical advice about adverse events. You are encouraged to
report adverse events related to Pfizer products by calling 1-800-438-1985 (U.S.
only). If you prefer, you may contact the U.S. Food and Drug Administration (FDA) directly.
Visit www.fda.gov/MedWatch or call 1-800-FDA-1088
.
LORBRENA®
is a registered trademark of Pfizer Inc.
|
|
|

|
|
|
|
© 2026
Pfizer Inc. All rights reserved. Pfizer
Privacy Policy
PP-LOR-USA-1168
|
|


