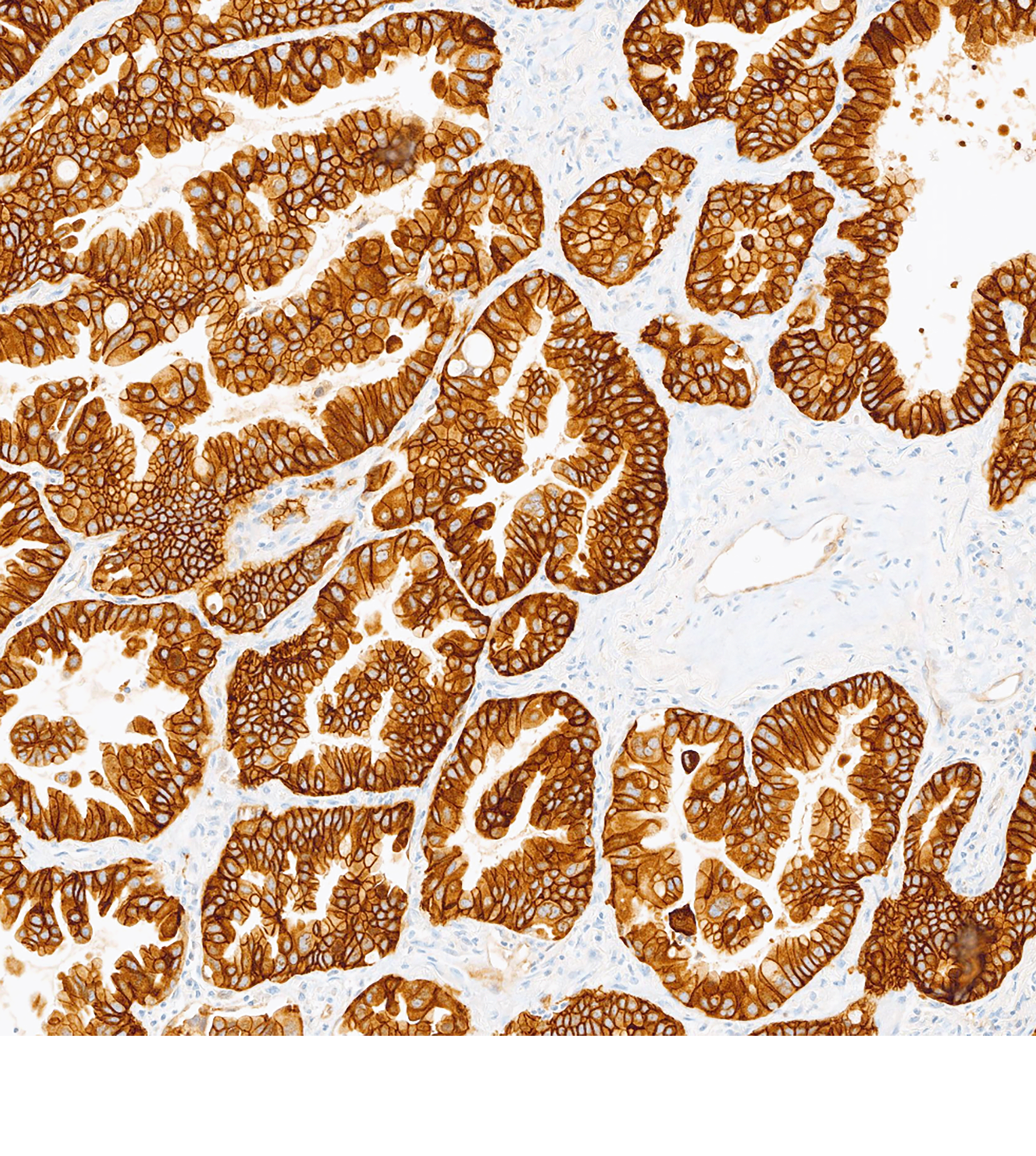
First and only c-Met targeted ADC for 2L+ advanced/metastatic NSq NSCLC with high c-Met protein overexpression1†
AbbVie is excited to announce that EMRELIS (telisotuzumab vedotin-tllv) is now FDA-approved
for 2L+ NSq NSCLC in patients with high c-Met protein overexpression [≥50% of tumor cells with strong (3+) staining].1†
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).1
Until now, there have been no approved therapies for c-Met protein overexpressed NSCLC.2 That changes with EMRELIS. For the first time, patients with high c-Met protein overexpressed NSq NSCLC have a treatment designed specifically for them.1
INDICATION
EMRELIS is indicated for the treatment of adult patients with locally advanced or metastatic, non‑squamous non‑small cell lung cancer (NSCLC) with high c-Met protein overexpression [≥50% of tumor cells with strong (3+) staining], as determined by an FDA-approved test, who have received a prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
SELECT IMPORTANT SAFETY INFORMATION
Warnings and Precautions Peripheral neuropathy, interstitial lung disease/pneumonitis, ocular surface disorders, infusion-related reactions, and embryo-fetal toxicity.
Adverse Reactions Serious adverse reactions occurred in 35% of patients. The most common adverse reactions (≥20%) were peripheral neuropathy, fatigue, decreased appetite, and peripheral edema.
The most common Grade 3 or 4 laboratory abnormalities (≥2%) were decreased lymphocytes, increased glucose, increased alanine aminotransferase, increased gamma glutamyl transferase, decreased phosphorus, decreased sodium, decreased hemoglobin, and decreased calcium.
Please see full Important Safety Information below.
IDENTIFY PATIENTS NOW
MET IHC testing is essential to identify patients who may be eligible for treatment with EMRELIS.1 Testing can be performed at diagnosis or thereafter (with archived tissue or re-biopsy).2‡
The VENTANA MET (SP44) RxDx Assay is a companion diagnostic to identify high c-Met protein overexpression.3†
VENTANA MET (SP44) RxDx Assay is indicated as an aid in identifying NSCLC patients eligible for treatment with EMRELIS for the indication and MET cutoff in accordance with the approved therapeutic product labeling. For more information, please refer to the official FDA approval and product labeling.
Learn more about how EMRELIS may help your patients
EMRELIS can cause peripheral neuropathy, including peripheral sensory neuropathy and peripheral motor neuropathy. In the safety population, peripheral neuropathy occurred in 51% of patients treated with EMRELIS, including Grade 3 in 11%. These adverse reactions included peripheral sensory neuropathy in 45% of patients and peripheral motor neuropathy in 9%. The median time to onset of peripheral neuropathy was 105 days (range: 1 to 472 days). Peripheral neuropathy led to permanent discontinuation of EMRELIS in 13% of patients. The median time to onset of peripheral neuropathy leading to treatment discontinuation was 249 days (range: 57 to 519 days). Of the 7 patients with motor neuropathy ongoing as of their last dose of EMRELIS, 6 had persistent Grade 1 or 2
symptoms 30 days after their last dose.
Monitor patients for signs and symptoms of new or worsening peripheral neuropathy such as hypoesthesia, hyperesthesia, paresthesia, a burning sensation, neuropathic pain, or muscle weakness. Withhold, reduce the dose, or permanently discontinue EMRELIS based on severity.
Interstitial Lung Disease/Pneumonitis
EMRELIS can cause severe, life-threatening, or fatal interstitial lung disease (ILD)/pneumonitis. In the safety population, ILD/pneumonitis occurred in 10% of patients treated with EMRELIS, including Grade 3 in 3% and Grade 4 in 0.6%. There were 3 fatal cases of ILD/pneumonitis in patients who received EMRELIS. The median time to onset of ILD/pneumonitis was 48 days (range: 23 to 85 days). ILD/pneumonitis led to permanent discontinuation of EMRELIS in 7% of patients. The median time to onset of ILD/pneumonitis leading to treatment discontinuation was 46 days (range: 23 to 85 days).
Advise patients to immediately report cough, dyspnea, fever, and/or any new or worsening respiratory symptoms. Monitor patients for signs and symptoms of ILD/pneumonitis. Withhold or permanently discontinue EMRELIS based on severity.
Ocular Surface Disorders
EMRELIS can cause ocular surface disorders, including blurred vision, visual impairment, keratitis, and dry eye. In the safety population, ocular surface disorders occurred in 25% of patients treated with EMRELIS. The most common ocular surface disorders were blurred vision (15%), keratitis (11%), and dry eye (5%). Grade 3 ocular surface disorders occurred in 1.2% of patients [blurred vision (1.2%), and keratitis (0.6%)]. The median time to onset of ocular surface disorders was 47 days (range: 1 to 319 days).
Monitor patients for ocular surface disorders during treatment with EMRELIS. Withhold EMRELIS and refer patients to an eye care professional for an ophthalmic examination and treatment for patients who develop Grade ≥2 ocular toxicity. Withhold or permanently discontinue EMRELIS based on severity.
Infusion-Related Reactions
EMRELIS can cause infusion-related reactions (IRR); signs and symptoms of IRR include dyspnea, flushing, chills, nausea, chest discomfort, and hypotension. The median time to onset of IRR was 28 days (range: 1 to 43 days). In the safety population, IRR occurred in 3% of patients treated with EMRELIS, including Grade 3 in 1.2% and Grade 4 in 0.6%. IRR led to permanent discontinuation of EMRELIS in 0.6% of patients.
Monitor patients for signs and symptoms of infusion reactions during EMRELIS infusion. Withhold, reduce the rate of infusion, or permanently discontinue EMRELIS based on severity. For patients who experience IRR, administer premedications prior to subsequent infusions.
Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, EMRELIS can cause fetal harm when administered to a pregnant woman. The small molecule component of EMRELIS, monomethyl auristatin E (MMAE), administered to rats caused adverse developmental outcomes, including embryo-fetal mortality and structural abnormalities, at exposures similar to those occurring clinically at the recommended dose.
Advise patients of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with EMRELIS and for 2 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with EMRELIS and for 4 months after the last dose.
Adverse Reactions
Serious adverse reactions occurred in 35% of patients. The most common adverse reactions (≥20%) were peripheral neuropathy, fatigue, decreased appetite, and peripheral edema.
The most common Grade 3 or 4 laboratory abnormalities (≥2%) were decreased lymphocytes, increased glucose, increased alanine aminotransferase, increased gamma glutamyl transferase, decreased phosphorus, decreased sodium, decreased hemoglobin, and decreased calcium.
Drug Interactions
Strong CYP3A Inhibitors: Concomitant use with EMRELIS may increase the area under the curve of MMAE. Monitor for increased risk of adverse reactions to EMRELIS.
Use in Specific Populations
Severe or Moderate Hepatic Impairment: Avoid the use of EMRELIS.
Lactation: Advise lactating women not to breastfeed during treatment with EMRELIS and for 1 month after the last dose.
Infertility: Based on findings from animal studies, EMRELIS may impair fertility in females and males.
INDICATION
EMRELIS is indicated for the treatment of adult patients with locally advanced or metastatic, non-squamous non-small cell lung cancer (NSCLC) with high c-Met protein overexpression [≥50% of tumor cells with strong (3+) staining], as determined by an FDA-approved test, who have received a prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
†High c-Met protein overexpression is defined as ≥50% of tumor cells with strong (3+) staining.1
‡Screening samples for patients enrolled in LUMINOSITY were collected from diagnosis through post-progression.2
2L=second line; ADC=antibody-drug conjugate; FDA=Food and Drug Administration; IHC=immunohistochemistry; NSCLC=non-small cell lung cancer; NSq=non-squamous.
REFERENCES:
1. EMRELIS [package insert]. North Chicago, IL: AbbVie Inc.; 2025. 2. Camidge DR, Bar J, Horinouchi H, et al. Telisotuzumab vedotin monotherapy in patients with previously treated c-Met protein–overexpressing advanced nonsquamous EGFR-wildtype non–small cell lung cancer in the Phase II LUMINOSITY Trial. J Clin Oncol.
2024;42(25):3000-3011. 3. Roche Diagnostics. VENTANA MET (SP44) RxDx Assay Method Sheet. Roche; 2025.
This email has been sent to you because we believe information about EMRELIS™ may be of value to you. If you no longer wish to receive emails about EMRELIS™, please unsubscribe.
We respect your privacy. For more information, please view our privacy notice.
Please DO NOT REPLY TO THIS MESSAGE. Contact us with questions or comments.
Mail written correspondence to:
AbbVie Customer Service Department 36M 1 North Waukegan Road North Chicago, IL 60064-6163
This content is sponsored by AbbVie, and ION Oncology Practice Network has not independently reviewed or verified the information provided by AbbVie.
This email was sent to karleigh.bard-marsh@cencora.com because you have an account with or are a valued partner of Cencora Specialty GPOs | ION Oncology Practice Network. If you no longer wish to receive these types of emails, please click here to unsubscribe or update your email preferences.